





La petición ahora está abierta a comentarios públicos.
Una petición ciudadana presentada a la Administración de Alimentos y Medicamentos de Estados Unidos (FDA) el 20 de enero de 2025 solicita la suspensión o revocación total de las vacunas contra la Covid-19 Comirnaty de Pfizer-BioNTech y Spikevax de Moderna .
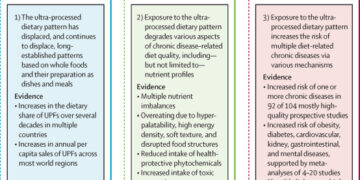
La petición alega violaciones de los procesos regulatorios, plantea serias preocupaciones de seguridad y finalmente concluye que las vacunas fueron “aprobadas ilegalmente”.
Presentada por la abogada Katie Ashby-Koppens de PJ O’Brien & Associates, la petición está encabezada por el ex abogado Julian Gillespie , junto con el director científico y fundador de Medicinal Genomics Kevin McKernan , la bióloga computacional Dra. Jessica Rose , el virólogo Dr. David Speicher y la consultora farmacéutica Maria Gutschi .

Deficiencias regulatorias
En el centro de la petición está la afirmación de que la FDA “de manera ilícita e ilegal” permitió a Pfizer y Moderna eludir las Evaluaciones Ambientales (EA) legalmente requeridas según la Parte 25 del Título 21 del CFR , un paso regulatorio crucial para los productos biológicos.
Las EA evalúan los riesgos potenciales para la salud humana y el medio ambiente, en particular para los productos clasificados como «terapias génicas». La petición sostiene que las vacunas de ARNm se ajustan a la definición de la FDA de productos de terapia génica, dado que utilizan material genético sintético para inducir efectos terapéuticos.
Al tratar estas vacunas como productos biológicos convencionales en lugar de terapias génicas, argumenta la petición, la FDA eludió procesos de evaluación más estrictos y negó la transparencia pública al omitir el período de comentarios requerido. Según la Sección 25.15(a), la ausencia de una evaluación previa (EA) adecuada por sí sola justifica el rechazo de una solicitud.
Además, la petición afirma que las vacunas fueron revisadas incorrectamente por el Comité Asesor de Vacunas y Productos Biológicos Relacionados (VRBPAC) en lugar del más apropiado Comité Asesor de Terapias Celulares, Tisulares y Génicas (CTGTAC) , que se especializa en evaluaciones de terapia genética.
Cabe destacar que la propia Moderna reconoció que su tecnología de ARNm está sujeta a la regulación de la terapia génica. En una presentación ante la Comisión de Bolsa y Valores (SEC) de 2020 , la compañía declaró: «Actualmente, la FDA considera el ARNm un producto de terapia génica».
Incluso la propia definición de la FDA sobre la terapia génica humana —“modificar o manipular la expresión de un gen o alterar las propiedades biológicas de las células vivas para uso terapéutico”— refleja la función de las vacunas de ARNm, sostiene la petición.
La contaminación del ADN como un problema de seguridad
La petición destaca serias preocupaciones de seguridad relacionadas con la contaminación del ADN en las vacunas de ARNm de Pfizer y Moderna.
Se citan múltiples estudios, incluido uno realizado en el propio laboratorio de White Oak de la FDA, que informa niveles de ADN residual entre 6 y 470 veces superiores al límite reglamentario de 10 nanogramos por dosis.
La petición señala que los fragmentos de ADN están encapsulados en nanopartículas lipídicas (junto con ARNm), lo que los protege de la detección inmunológica y permite que el ADN extraño ingrese a las células humanas, incluidos sus núcleos.
Este proceso pone en riesgo la integración genómica y la autorreplicación, lo que podría dar lugar a cánceres como la leucemia y alteraciones genéticas hereditarias.
Otras preocupaciones giran en torno a la presencia de secuencias promotoras del virus simio 40 (SV40) encontradas en viales de vacunas, que pueden unirse a proteínas supresoras de tumores como p53, neutralizando la capacidad del cuerpo para prevenir crecimientos cancerosos.
Los riesgos se extienden al daño transgeneracional, ya que el ADN sintético podría afectar las células de la línea germinal y causar alteraciones del desarrollo, abortos espontáneos o malformaciones.
Retención de datos
Se acusa a Pfizer de retener información sobre las secuencias promotoras del SV40 en sus mapas de ADN plasmídico, información que es requerida para la evaluación de la FDA según 21 CFR 601.2.
La petición alega que la omisión de esta información comprometió la capacidad de la FDA de evaluar completamente la seguridad de las vacunas.
Además, al no clasificar los productos como “terapias genéticas”, se alega que la FDA violó la confianza pública, eludiendo las leyes de transparencia y privando a las personas de la oportunidad de tomar decisiones informadas.
Esfuerzos previos
Esta no es la primera vez que se cuestionan las decisiones regulatorias sobre las vacunas contra la Covid-19.
En mayo de 2021, Robert Kennedy Jr. y Meryl Nass solicitaron la revocación de las Autorizaciones de Uso de Emergencia (EUA) para las vacunas contra la COVID-19, alegando problemas de seguridad. La FDA denegó la solicitud, alegando que no existía fundamento para la revocación.
En julio de 2021, Peter Doshi, editor senior de BMJ, y sus colegas solicitaron retrasar la aprobación total de cualquier vacuna contra la COVID-19, enfatizando la necesidad de más datos de seguridad a largo plazo. Esta petición también fue denegada por la FDA.
El abogado Aaron Siri, representante de la Red de Acción por el Consentimiento Informado (ICAN), presentó posteriormente una petición para revocar las autorizaciones de uso de emergencia (EUA) para las vacunas contra la COVID-19 en niños pequeños, planteando preocupaciones de seguridad similares. Esta solicitud también fue rechazada.
¿Qué sigue?
Los peticionarios exigen que la FDA suspenda inmediatamente las aprobaciones de vacunas, realice pruebas independientes de las existencias de vacunas retenidas y revise los umbrales de seguridad para los contaminantes del ADN sintético .
“La administración continua de estos productos sin abordar su contaminación con ADN sintético expone a millones de personas a riesgos de salud evitables, incluidos el cáncer, la inestabilidad genómica y el daño genético transgeneracional”, concluye la petición.
Según la ley federal de Estados Unidos, la FDA debe responder a la petición dentro de 180 días.
La petición , presentada bajo el número de expediente: FDA-2025-P-0335 en regulation.gov , está abierta a comentarios públicos y está pendiente la respuesta oficial de la FDA.
El resultado de esta petición servirá como una prueba crucial de la voluntad de la FDA de reevaluar su enfoque de la supervisión de las vacunas en medio de un creciente escrutinio, especialmente con un nuevo liderazgo en el horizonte.














{kind=link}